Supplemental: Diagnosing Unstable Systems
Overview
Teaching: 0 min
Exercises: 0 minQuestions
FIXME
Objectives
This episode is still work-in-progress.
FIXME
It is not uncommon for Molecular Dynamics simulation to become unstable for various reasons, resulting certain numerical algorithms to break and the simulation to crash. This is often referred to as the simulation is “blowing up” and especially inexperienced users often have difficulties with dealing with this.
This section provides some tips about what can be checked and how one can proceed.
When does the crash occur?
During Energy Minimization
If the crash happens during energy minimization, then it is quite likely that there are atomic clashes that are so severe that the minimization cannot resolve. Note down the atom-number that has the largest force and use it as a start for your investigation.
Other possibilities are that there is an error in the topology or a severe error in the MD-parameters (e.g. are the cut-offs specified in the correct unit – Å vs nm).
When using GROMACS
When the GROMACS energy minimization is executed with the -v option, it will print information about which atom has the larges force (Fmax) acting on it. e.g.:
Steepest Descents:
Tolerance (Fmax) = 1.00000e+03
Number of steps = 50000
Step= 0, Dmax= 1.0e-02 nm, Epot= -4.52720e+05 Fmax= 2.48664e+05, atom= 710
Step= 1, Dmax= 1.0e-02 nm, Epot= -4.63759e+05 Fmax= 8.99042e+04, atom= 710
Step= 2, Dmax= 1.2e-02 nm, Epot= -4.79680e+05 Fmax= 2.77594e+04, atom= 1046
Step= 3, Dmax= 1.4e-02 nm, Epot= -5.00649e+05 Fmax= 9.51331e+03, atom= 24272
Step= 4, Dmax= 1.7e-02 nm, Epot= -5.21439e+05 Fmax= 9.13588e+03, atom= 710
Step= 5, Dmax= 2.1e-02 nm, Epot= -5.32010e+05 Fmax= 1.79030e+04, atom= 710
When using AMBER
FIXME
When using NAMD
FIXME
During initial NVT Simulation
- Check whether the Energy minimization was successful, if not go back and troubleshoot from that point.
- Did you generate an initial set of velocities based on the Maxwell-Boltzmann distribution for the target temperature? If not you are heating up the system from 0K, which not only takes longer, but also could be problematic with second-order thermostats (see below).
- Check whether you are using a suitable method for temperature control at this point. Extended ensemble (second-order) thermostats like Nosé-Hoover will approach the target temperature with a dampened oscillation. If the starting temperature is far from the target temperature, it will take many oscillations and the system can become unstable. During the relaxation phase, it is recommended to use a first-order thermostat like Berendsen or Bussi’s stochastic velocity rescaling.
FIXME: Is the Langevin thermostat also prone to fail during relaxation phase?
During initial NPT Simulation
FIXME
Using VMD to Inspect Atomic Clashes
If you have identified some atoms as “suspects” (e.g. atoms mentioned in LINCS, SHAKE or SETTLE warnings, or having very large forces during energy minimization), it can be helpful to visualize the vicinity around those “suspects” with a molecular viewer like VMD. The same strategy can also be applied with other molecular viewers.
-
Open the initial coordinate file (starting structure) and the trajectory in VMD and advance to the timesteps closet to the crash.
If your MD-package has saved coordinate files for the time-steps leading up to the crash, you can load these instead. -
Open the “Grahical Representations” window (Graphics > Representations…) and create a new representation: Create Rep..
-
Select the “suspect” atom (e.g.
serial = 710) and set it to style VDW so that it stands out. -
Create another representation (Create Rep.) in which you you select residues within a certain distance (e.g. between 5 and 10 Å). around the central atom and set a style (e.g. Lines or CPK). The selection command for selecting all residues that have atoms within 7 Å around atom number
710is:same resid as (within 7 of serial = 710). -
Toggle off the original representation (selection “all”) by double-clicking on that line in the list. The result should look like this:
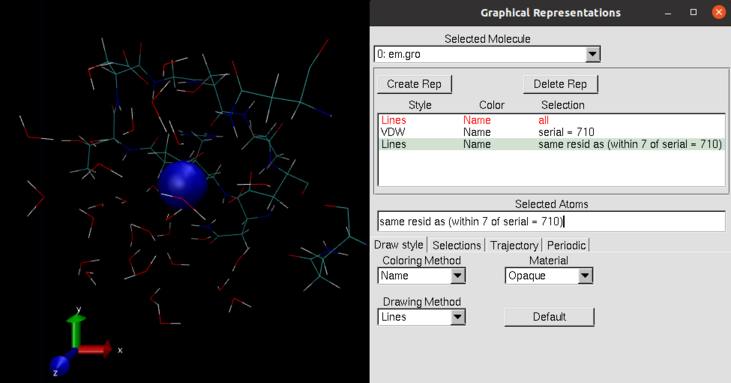
-
Set Mouse to “Pick” and open the “Graphics > Labels…” window, so that you can identify the atom/residue number of the offending atoms/molecules.
-
If you are looking at a trajectory, you can also step one-by-one through a few adjacent frames to get a sense how the system has been evolving leading up to the crash. Is there an atom moving very fast and hitting a series of other atoms that were flagged by warnings?
-
You may need to repeat this with several atoms that you have on your list of suspects.
Additional Resources
- GROMACS Manual
Key Points
FIXME