Preparation and simulation of membrane and membrane-protein systems
Overview
Teaching: 30 min
Exercises: 5 minQuestions
How to prepare a membrane simulation system?
How to pack a protein in a lipid bilayer?
Objectives
Prepare a membrane simulation system
Pack a protein in a lipid bilayer
Creating simulation systems with packmol-memgen
AMBER Lipid force fields
- Lipid21 is the main membrane force field
- Lipids are modeled as polymers composed of a headgroup and acyl tails
Known Issues: Using MC barostat with hard LJ cutoff is known to cause bilayer deformation. It is recommended to use an LJ force switch when running simulations with the MC barostat.Gomez, 2021
Useful links
- There is a great deal of information provided in Dickson, 2022 regarding best practices when using AMBER for lipid simulations.
- Full list of supported lipids is in Section 3.4 of Amber22 manual
Creating a membrane-only simulation system
- What lipids are available?
cd ~/workshop_amber/example_06
module purge
module load StdEnv/2023 ambertools
packmol-memgen --available_lipids
- To see all available lipids use
--available_lipids_all, but the list will have thousands of items!
#!/bin/bash
#SBATCH --mem-per-cpu=2000M
#SBATCH --time=3:00:00
module purge
module load StdEnv/2023 ambertools/23
packmol-memgen \
--lipids DOPE:DOPG \
--ratio 3:1 \
--distxy_fix 50 \
--parametrize
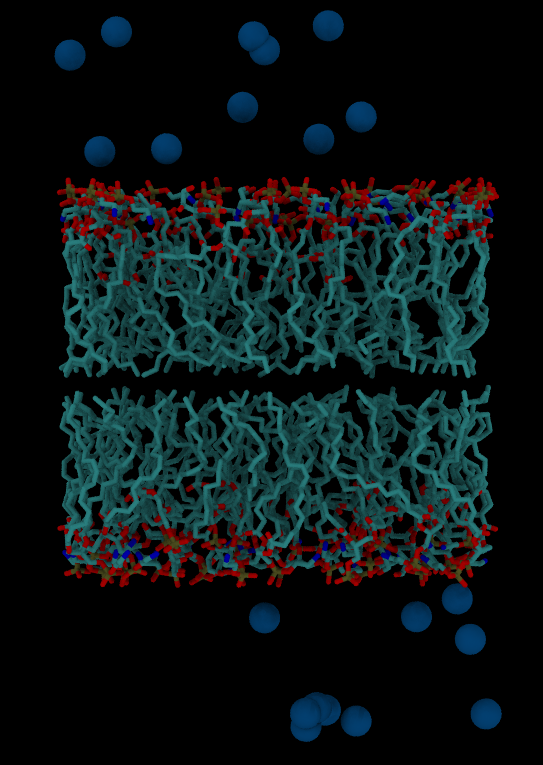
- if the option –parametrize is given the solvated system is bilayer_only_lipid (top, crd, pdb)
- without –parametrize the solvated system is bilayer_only.pdb
- Multiple bilayers can be generated by repeating corresponding flags.
- Bilayers with different leaflet composition can be generated:
--lipids PSM:POPC:POPS:POPE//PSM:POPC:POPS:POPE \
--ratio 21:19:1:7//2:6:15:25 \
Example illustrating the building of a bilayer where leaflets consist of different types of lipids is in the section “Hands-on 1: Packing a complex mixture of different lipid species into a bilayer and simulating it”.
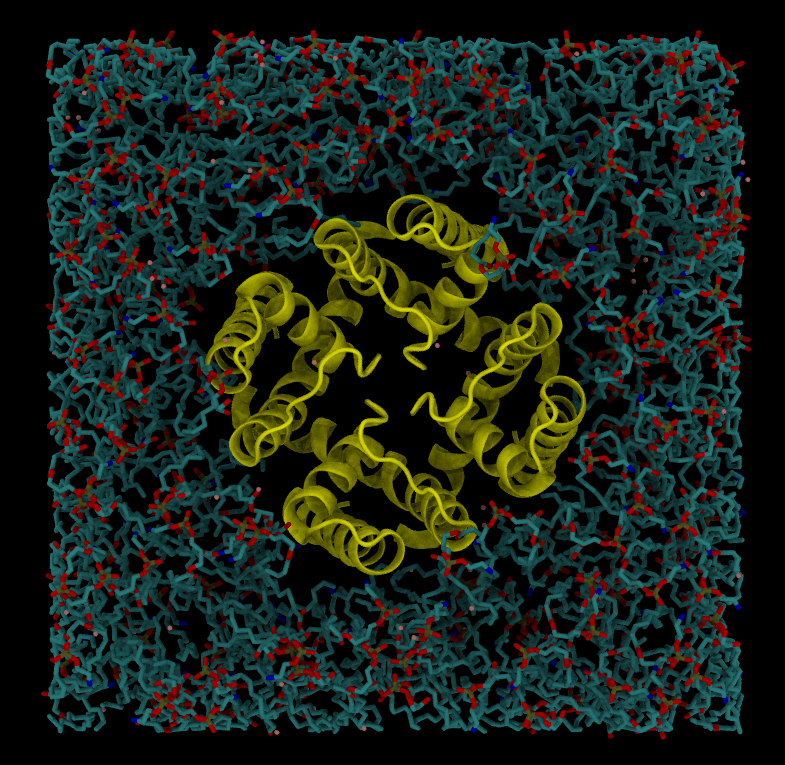
Embedding a protein into a bilayer
We will use PDB file 6U9P (wild-type MthK pore in ~150 mM K+) for this exercise.
- Use PPM server to orient a protein. PPM server will also take care of assembling the complete tetrameric pore.
- Use vmd to remove ligands and conformers B.
module purge
module load StdEnv/2023 vmd
vmd
mol new 6U9Pout.pdb
set sel [atomselect top "protein and not altloc B"]
$sel writepdb 6U9P-clean.pdb
quit
- Embed the protein into the lipid bilayer
#!/bin/bash
#SBATCH --mem-per-cpu=2000M
#SBATCH --time=3:00:00
module purge
module load StdEnv/2023 ambertools/23
packmol-memgen \
--pdb 6U9P-clean.pdb \
--lipids DOPE:DOPG \
--ratio 3:1 \
--preoriented \
--parametrize
- To create topology.
--parametrize - The default protein force field is FF14SB, water model TIP3P
- To use a different force field:
--fprot ff19SB --ffwat opc --gaff2 - To add salt (default K+, 0.15M):
--salt - To make bilayer patch larger:
--dims 95 95 85
Links to advanced AMBER tutorials
- Placing waters and ions using 3D-RISM and MOFT
- Building a Membrane System with PACKMOL-Memgen
- Minimizing and Equilibrating a packed membrane system
- Setup and simulation of a membrane protein with AMBER Lipid21 and PACKMOL-Memgen.
Key Points