Hands-on 1: Packing a complex mixture of different lipid species into a bilayer and simulating it
Overview
Teaching: 30 min
Exercises: 5 minQuestions
How to pack a complex mixture of different lipid species into a bilayer?
How to minimize energy?
How to heat up a simulation system?
How to equilibrate a simulation system?
Objectives
Prepare a lipid bilayer
Energy minimize, heat up and equilibrate the system
Introduction
This episode guides you through the process of building a bilayer, preparing simulation input files, minimizing energy, equilibrating the system, and running an equilibrium molecular dynamics simulation. You will need to follow a number of steps to complete the tutorial. If you are already familiar with some of these topics, you can skip them and focus on the ones you don’t know about.
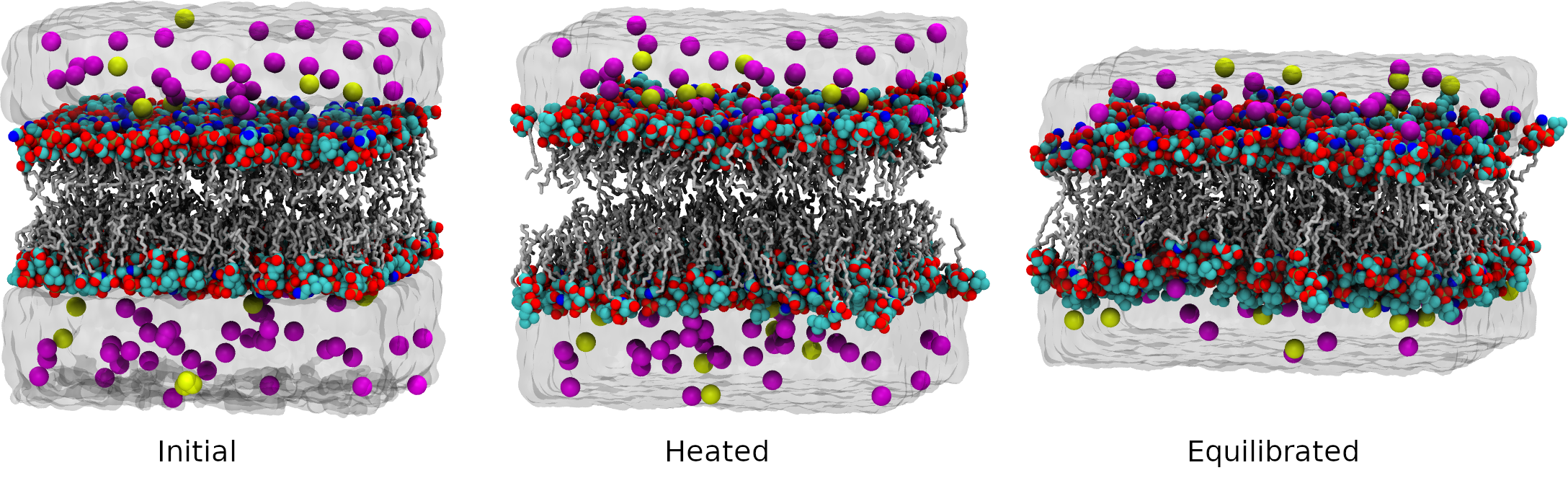
1. Generating a bilayer by packing lipids together.
The packmol-memgen program allows the creation of asymmetric bilayers with leaflets composed of different lipid species.
Bilayer asymmetry is a common feature of biological membranes. For example, the composition of the phospholipids in the erythrocyte membrane is asymmetric. Here is an article that reviews this topic in detail: Interleaflet Coupling, Pinning, and Leaflet Asymmetry—Major Players in Plasma Membrane Nanodomain Formation.
Asymmetric lipid bilayer can be generated by the following submission script:
#!/bin/bash
#SBATCH --mem-per-cpu=4000M
#SBATCH --time=1:00:00
#SBATCH --cpus-per-task=1
module purge
module load StdEnv/2020 gcc/9.3.0 openmpi/4.0.3 ambertools/23
rm -f bilayer* *.log
packmol-memgen \
--lipids PSM:POPC:POPS:POPE//PSM:POPC:POPS:POPE \
--salt --salt_c Na+ --salt_a Cl- --saltcon 0.25 \
--dist_wat 25 \
--ratio 21:19:1:7//2:6:15:25 \
--distxy_fix 100 \
--parametrize
The job takes about 30 minutes. It generates several output files:
- AMBER topology bilayer_only_lipids.top
- AMBER coordinates bilayer_only_lipids.crd
- PDB-formatted system bilayer_only_lipids.pdb
By default LIPID17, ff14SB and TIP3P force fields are used.
2. Using AMBER force fields not available natively in packmol-memgen.
Packmol-memgen offers several recent AMBER force fields that can be used to parameterize a bilayer. However, sometimes it is necessary to use force fields that are not readily available in packmol-memgen. The parameterization of a prepared bilayer with tleap directly is a way to achieve this. To use tleap you will need to provide an input file that is in pdb format compatible with AMBER.
Even though packmol-memgen creates a pdb file (bilayer_only_lipids.pdb), you should use it with caution. It may not work properly with some molecules. PSM is an example. When saving pdb file containing this type of lipids packmol-memgen erroneously adds TER record between the lipid headgroup (SPM) and second acyl tail (SA). This splits PSM molecule in two: PA+SPM and SA.
To illustrate the parameterization process, we will create a pdb file from AMBER topology and coordinate files, and then parameterize the bilayer using the polarizable water OPC3-pol not yet available natively in packmol-memgen.
Begin by loading the ambertools/23 module and starting the Python interpreter:
module load StdEnv/2020 gcc/9.3.0 openmpi/4.0.3 ambertools/23
python
In the python prompt enter the following commands:
import parmed
amber = parmed.load_file("bilayer_only_lipid.top", "bilayer_only_lipid.crd")
amber.save("bilayer-lipid21-opc3pol.pdb")
quit()
We only need to know one more thing before we can proceed: the dimensions of the periodic box. Box information can be extracted from the file bilayer_only_lipid.top:
grep -A2 BOX bilayer_only_lipid.top
...
%FORMAT (5E168)
9.00000000E+01 1.03536600E+02 1.03744300E+02 9.97203000E+01
The last three numbers refer to the size of the box in the X, Y, and Z axes. We can now parameterize the system using the Lipid21 force field and OPC3-Pol polarizable water as follows:
module load StdEnv/2020 gcc/9.3.0 openmpi/4.0.3 ambertools/23
tleap -f <(cat << EOF
source leaprc.water.opc3pol
source leaprc.lipid21
sys=loadpdb bilayer-lipid21-opc3pol.pdb
set sys box {103.5 103.7 99.7}
set default nocenter on
saveamberparm sys bilayer.parm7 bilayer.rst7
quit
EOF)
Don’t forget to replace {103.5 103.5 99.9} with your own numbers!
3. Energy minimization
A system must be optimized in order to eliminate clashes and prepare it for molecular dynamics.
To run energy minimization we need three files:
- Molecular topology bilayer.parm7
- Initial coordinates bilayer.rst7
- Simulation input file minimization.in
First, we create a directory for energy minimization job.
mkdir 1-minimization && cd 1-minimization
Here is an input file for minimization (minimization.in) that you can use.
Minimization
&cntrl
imin=1, ! Minimization
ntmin=3, ! Steepest Descent
maxcyc=1000, ! Maximum number of cycles for minimization
ntpr=10, ! Print to mdout every ntpr steps
ntwr=1000, ! Write a restart file every ntwr steps
cut=10.0, ! Nonbonded cutoff
/
Using the script below, submit a minimization job.
#!/bin/bash
#SBATCH --mem-per-cpu=4000
#SBATCH --ntasks=10
#SBATCH --time=1:00:00
module purge
module load StdEnv/2020 gcc/9.3.0 openmpi/4.0.3 ambertools/23
srun sander.MPI -O -i minimization.in \
-o minimization.out \
-p ../bilayer.parm7 \
-c ../bilayer.rst7 \
-r minimized.rst7
When the optimization job is completed, the output file minimized.rst7 will contain the optimized coordinates. Minimization takes about 40 minutes.
4. Heating
Start with creating a directory for energy heating job.
cd ..
mkdir 2-heating && cd 2-heating
Our optimized simulation system does not move yet, there is no velocities. Our next step is to heat it up to room temperature. The system can be heated in a variety of ways with different initial temperatures and heating rates. It’s important to warm up the system gently, avoiding strong shocks that can create conformations that aren’t physiological. A typical protocol is described in Amber tutorial Relaxation of Explicit Water Systems.
We will start the system at the low temperature of 100K and heat it up to 300K over 20 ps of simulation time at constant pressure. At this step we use weak coupling thermostat and barostat with both time constants set to 5 ps.
Here is how the simulation input file heating.in looks like:
Heating
&cntrl
imin=0, ! Molecular dynamics
ntx=1, ! Read coordinates with no initial velocities
ntc=2, ! SHAKE on for bonds with hydrogen
ntf=2, ! No force evaluation for bonds with hydrogen
tol=0.0000001, ! SHAKE tolerance
nstlim=20000, ! Number of MD steps
ntt=1, ! Berendsen thermostat
tempi=100, ! Initial temperature
temp0=300, ! Target temperature
tautp=5.0, ! Time constant for heat bath coupling
ntpr=100, ! Write energy info every ntwr steps
ntwr=10000, ! Write restart file every ntwr steps
ntwx=2000, ! Write to trajectory file every ntwx steps
dt=0.001, ! Timestep
ntb=2, ! Constant pressure periodic bc.
barostat=1, ! Berendsen barostat
taup=5, ! Pressure relaxation time
ntp=3, ! Semiisotropic pressure scaling
csurften=3, ! Constant surface tension with interfaces in the xy plane
cut=10.0, ! Nonbonded cutoff
/
Submit job using the following script:
#!/bin/bash
#SBATCH --mem-per-cpu=4000M
#SBATCH --gpus-per-node=v100:1
#SBATCH --time=1:00:00
module purge
module load StdEnv/2020 gcc/9.3.0 cuda/11.4 openmpi/4.0.3 amber/20.12-20.15
pmemd.cuda -O -i heating.in \
-o heating.out \
-p ../bilayer.parm7 \
-c minimized.rst7 \
-r heated.rst7
A minute is all it takes for this job to be completed.
5. The first phase of the equilibration process.
The second step of relaxation will maintain the system at 300 K over 100 ps of simulation time at a constant pressure, allowing the box density to relax. The initial density is too low because there are gaps between lipids an water and lipids are not perfectly packed.
As the simulation parameters are the same as on the previous step, you might wonder why we did not simply continue with the previous step? It is necessary to restart because the box size changes rapidly from its original size, and a running simulation cannot handle such large changes (GPU code does not reorganize grid cells at runtime). Restarting simulation will generate new grid cells and allow the simulation to continue.
Simulation input file equilibration.in:
Equilibration 1
&cntrl
imin=0, ! Molecular dynamics
irest=1, ! Restart
ntx=5, ! Read positions and velocities
ntc=2, ! SHAKE on for bonds with hydrogen
ntf=2, ! No force evaluation for bonds with hydrogen
tol=0.0000001, ! SHAKE tolerance
nstlim=200000, ! Number of MD steps
ntt=1, ! Berendsen thermostat
temp0=300, ! Set temperature
tautp=5.0, ! Time constant for heat bath coupling
ntpr=100, ! Write energy info every ntwr steps
ntwr=10000, ! Write restart file every ntwr steps
ntwx=2000, ! Write to trajectory file every ntwx steps
dt=0.001, ! Timestep (ps)
ntb=2, ! Constant pressure periodic bc.
barostat=1, ! Berendsen barostat
taup=5, ! Pressure relaxation time
ntp=3, ! Semiisotropic pressure scaling
csurften=3, ! Constant surface tension with interfaces in the xy plane
fswitch=8.0, ! Force switching
cut=10.0, ! Nonbonded cutoff
/
Submission script
#!/bin/bash
#SBATCH --mem-per-cpu=4000M
#SBATCH --gpus-per-node=v100:1
#SBATCH --time=1:00:00
module purge
module load StdEnv/2020 gcc/9.3.0 cuda/11.4 openmpi/4.0.3 amber/20.12-20.15
pmemd.cuda -O -i equilibration.in \
-o eq-1.out \
-p ../bilayer.parm7 \
-c ../2-heating/heated.rst7 \
-r eq-1.rst7
This job completes in about 5 minutes.
Examine energy components
log=eq-1.out
cpptraj << EOF
readdata $log
writedata energy.dat $log[Etot] $log[TEMP] $log[PRESS] $log[VOLUME] time 0.1
EOF
6. The second phase of the equilibration process.
Now density is mostly relaxed and we can apply temperature and pressure coupling parameters appropriate for a production run. Temperature and pressure coupling methods are changed to produce the correct NPT ensemble. To speed up simulation, we use a longer time step. The relaxation of a bilayer may take many nanoseconds. We simulate only for 1,000,000 steps because box dimensions are still far from equilibration and restart will likely be necessary to reorganize grid cells.
Simulation input file equilibration-2.in:
Equilibration 2
&cntrl
imin=0, ! Molecular dynamics
irest=1 ! Restart
ntx=5, ! Read positions and velocities
ntc=2, ! SHAKE on for bonds with hydrogen
ntf=2, ! No force evaluation for bonds with hydrogen
tol=0.0000001, ! SHAKE tolerance
nstlim=1000000, ! Number of MD steps
ntt=3, ! Langevin thermostat
temp0=300, ! Target temperature
gamma_ln=2.0, ! Collision frquency
ntpr=100, ! Write energy info every ntwr steps
ntwr=10000, ! Write restart file every ntwr steps
ntwx=10000, ! Write to trajectory file every ntwx steps
dt=0.002, ! Timestep (ps)
ntb=2, ! Constant pressure periodic bc.
barostat=1, ! Berendsen barostat
taup=1.0, ! Pressure relaxation time
ntp=3, ! Semiisotropic pressure scaling
csurften=3, ! Constant surface tension with interfaces in the xy plane
fswitch=10.0, ! Force switching from 10 to 12 A
cut=12.0, ! Nonbonded cutoff
/
Submission script
#!/bin/bash
#SBATCH --mem-per-cpu=4000M
#SBATCH --gpus-per-node=v100:1
#SBATCH --time=6:00:00
module purge
module load StdEnv/2020 gcc/9.3.0 cuda/11.4 openmpi/4.0.3 amber/20.12-20.15
pmemd.cuda -O -i equilibration-2.in \
-o eq-2.out \
-p ../bilayer.parm7 \
-c ../3-equilibration/eq-1.rst7 \
-r eq-2.rst7
Center trajectory using the bilayer COM
trajin ../bilayer.rst7
trajin ../2-heating/heated.rst7
trajin ../4-equilibration/eq-4.rst7
center :SPM,PA,PC,PS
image
trajout mdcrd.xtc
go
quit
Delete TER records between residues SPM and SA in bilayer_only_lipid.pdb using shell commands.
This exercise is optional for those who wish to improve their efficiency when working with text files. Using shell commands at an advanced level is the focus of the exercise.
Solution
The following command removes all erroneous TER records:
grep -n "H22 SPM " bilayer_only_lipid.pdb | \ cut -f1 -d: | \ awk '{print $0 + 1}' | \ awk -F, 'NR==FNR { nums[$0]; next } !(FNR in nums)' \ - bilayer_only_lipid.pdb > bilayer_only_lipid_fixed.pdbLet’s break it down:
grep -n "H22 SPM "finds all lines containing “H22 SPM “ and prints their line numbers followed by the line content.cut -f1 -d:selects the first field containing the line numberawk '{print $0 + 1}increments this number, after this command we have a list of lie numbers we want to deleteawk -F, 'NR==FNR { nums[$0]; next } !(FNR in nums)' - bilayer_only_lipid.pdbtakes the list of line numbers from the pipe and deletes lines with matching numbers from the file bilayer_only_lipid.pdbIf this command is executed, it will remove the line following the last atom of each SPM residue, effectively deleting the erroneous TER records.
Key Points