Force Fields and Interactions
Overview
Teaching: 30 min
Exercises: 0 minQuestions
What is Molecular Dynamics and how can I benefit from using it?
What is a force field?
What mathematical energy terms are used in biomolecular force fields?
Objectives
Understand strengths and weaknesses of MD simulations
Understand how the interactions between particles in a molecular dynamics simulation are modeled
Introduction
- Atoms and molecules interact with each other.
- We carry out molecular modeling by following and analyzing dynamic structural models in computers.
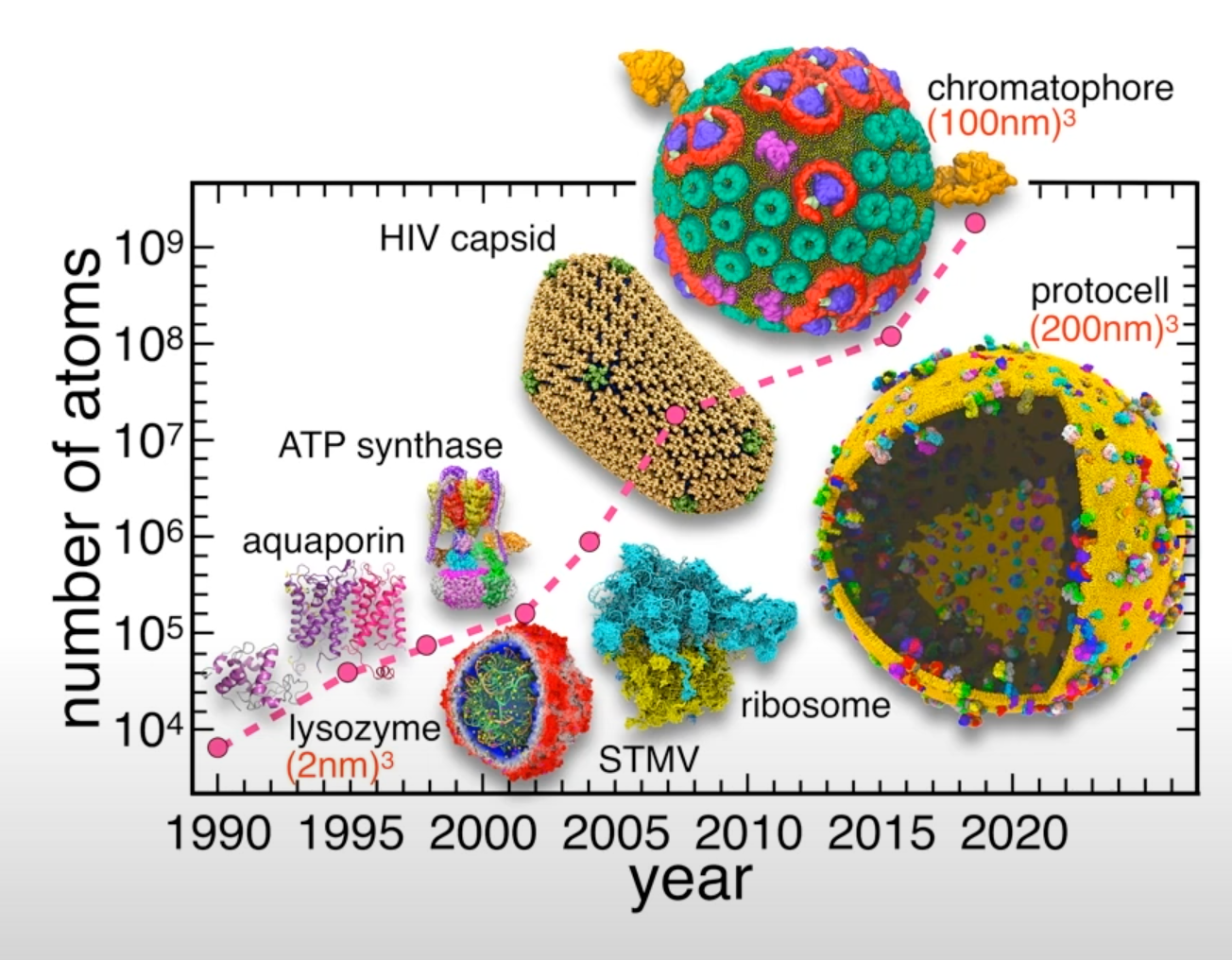 Figure from: Scalable molecular dynamics on CPU and GPU architectures with NAMD
Figure from: Scalable molecular dynamics on CPU and GPU architectures with NAMD
- The size and the length of MD simulations have been recently vastly improved.
- Longer and larger simulations allow us to tackle a wider range of problems under a wider variety of conditions.
Recent example - simulation of the whole SARS-CoV-2 virion
- System size: 304,780,149 atoms, 350 Å × 350 Å lipid bilayer, simulation time 84 ns
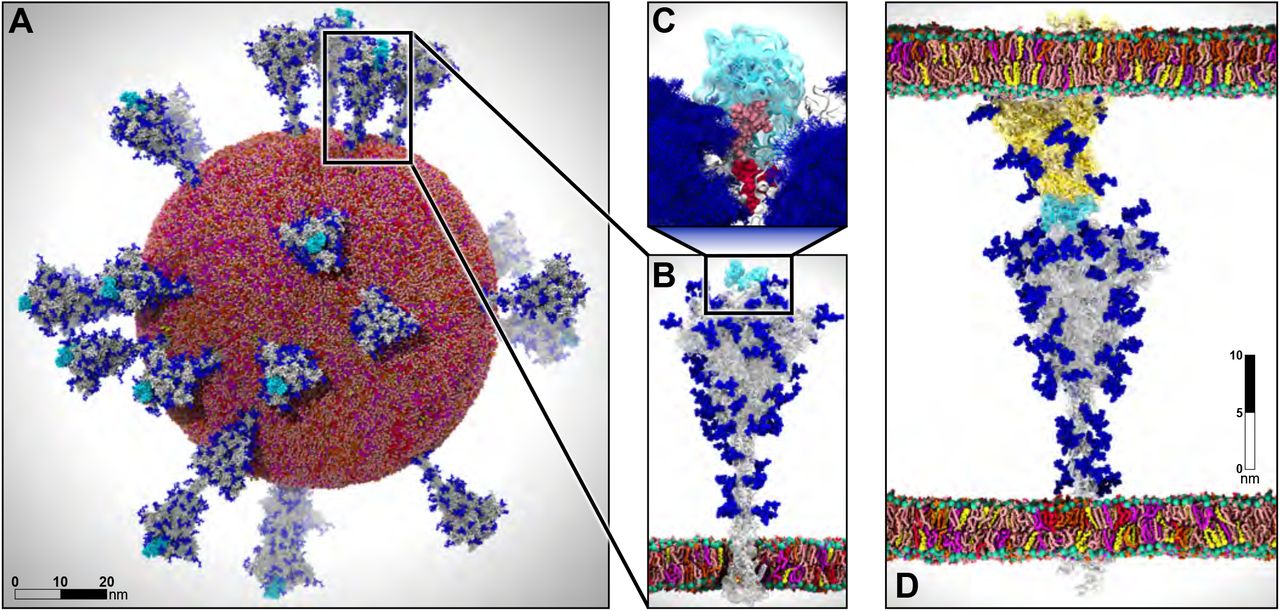 Figure from AI-Driven Multiscale Simulations Illuminate Mechanisms of SARS-CoV-2 Spike Dynamics
Figure from AI-Driven Multiscale Simulations Illuminate Mechanisms of SARS-CoV-2 Spike Dynamics
- Showed that spike glycans can modulate the infectivity of the virus.
- Characterized interactions between the spike and the human ACE2 receptor.
- Used ML to identify conformational transitions between states and accelerate conformational sampling.
Goals of the Workshop
- Introduce you to the method of molecular dynamics simulations.
- Guide you to using various molecular dynamics simulation packages and utilities.
- Teach how to use Digital Research Alliance of Canada (Alliance) clusters for system preparation, simulation and trajectory analysis.
The focus will be on reproducibility and automation by introducing scripting and batch processing.
The theory behind the method of MD.
Force Fields
- Understanding complex biological phenomena requires simulations of large systems for a long time window.
- The forces acting between atoms and molecules are very complex.
- Very fast method of evaluations molecular interactions is needed to achieve these goals.
Interactions are approximated with a simple empirical potential energy function.
- The potential energy function allows calculating forces: $ \vec{F}=-\nabla{U}(\vec{r}) $
- With the knowledge of the forces acting on an object, we can calculate how the position of that object changes over time: $ \vec{F}=\frac{d\vec{p}}{dt} $.
- Advance system with very small time steps assuming the velocities don’t change.
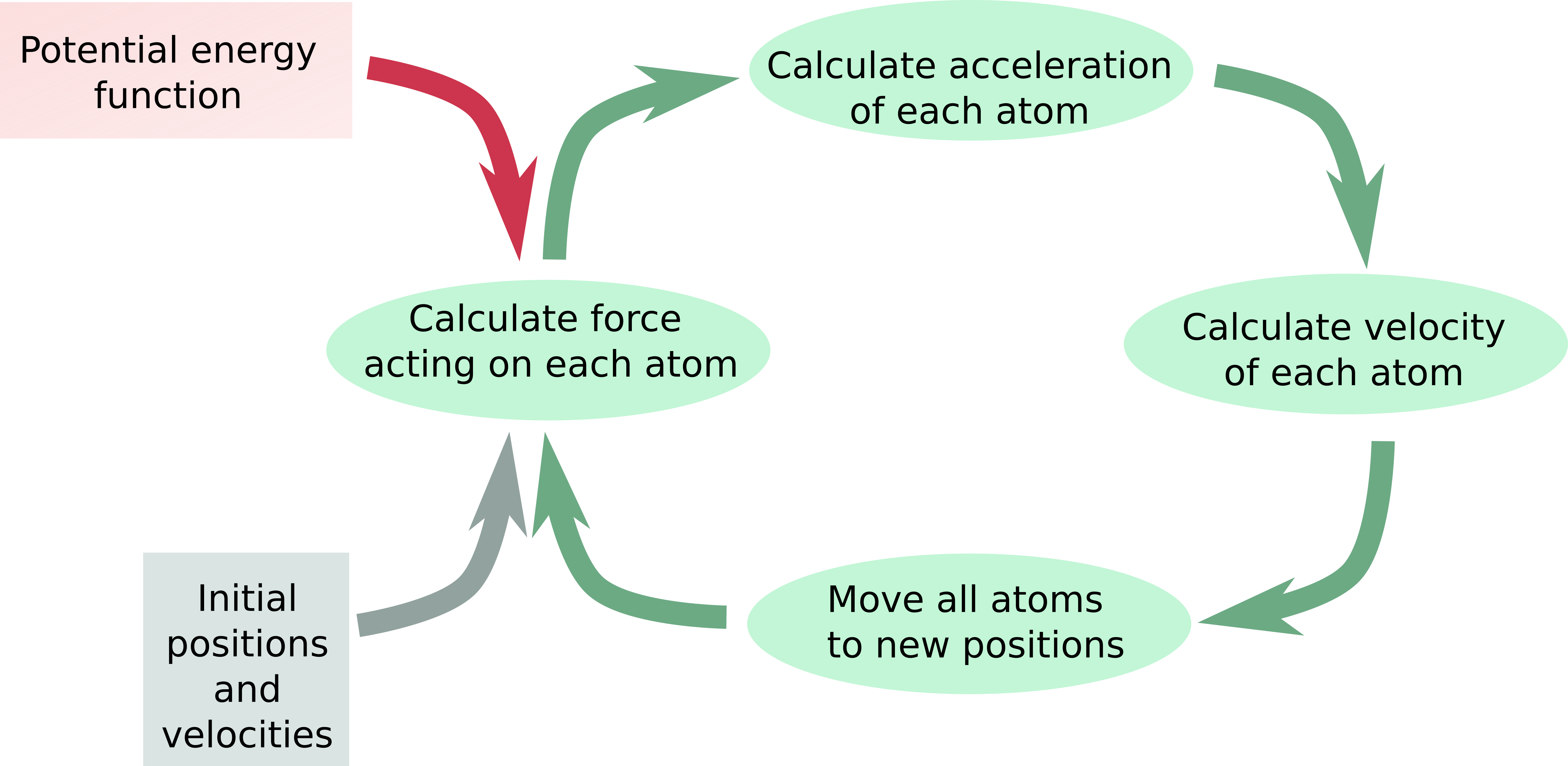 Flow diagram of MD simulation
Flow diagram of MD simulation
A force field is a set of empirical energy functions and parameters used to calculate the potential energy U as a function of the molecular coordinates.
- Potential energy function used in MD simulations is composed of non-bonded and bonded interactions:
$U(\vec{r})=\sum{U_{bonded}}(\vec{r})+\sum{U_{non-bonded}}(\vec{r})$
- Only pairwise interactions are considered.
Classification of force fields.
Class 1 force fields.
- Dynamics of bond stretching and angle bending is described by simple harmonic motion (quadratic approximation)
- Correlations between bond stretching and angle bending are omitted.
Examples: AMBER, CHARMM, GROMOS, OPLS
Class 2 force fields.
- Add anharmonic cubic and/or quartic terms to the potential energy for bonds and angles.
- Contain cross-terms describing the coupling between adjacent bonds, angles and dihedrals.
Class 3 force fields.
- Explicitly add special effects of organic chemistry such as polarization, stereoelectronic effects, electronegativity effect, Jahn–Teller effect, etc.
Examples of class 3 force fields are: AMOEBA, DRUDE
Energy Terms of Biomolecular Force Fields
Non-Bonded Terms
- Describe non-electrostatic and electrostatic interactions between all pairs of atoms.

- Non-electrostatic potential energy is most commonly described with the Lennard-Jones potential.
The Lennard-Jones potential
- Approximates the potential energy of non-electrostatic interaction between a pair of non-bonded atoms or molecules:
$V_{LJ}(r)=\frac{C12}{r^{12}}-\frac{C6}{r^{6}}$
- The \(r^{-12}\) term approximates the strong Pauli repulsion originating from overlap of electron orbitals.
- The \(r^{-6}\) term describes weaker attractive forces acting between local dynamically induced dipoles in the valence orbitals.
- The too steep repulsive part often leads to an overestimation of the pressure in the system.

- The LJ potential is commonly expressed in terms of the well depth \(\epsilon\) and the van der Waals radius \(\sigma\):
$V_{LJ}(r)=4\epsilon\left[\left(\frac{\sigma}{r}\right)^{12}-\left(\frac{\sigma}{r}\right)^{6}\right]$
- Relation between C12, C6, \(\epsilon\) and \(\sigma\):
$C12=4\epsilon\sigma^{12},C6=4\epsilon\sigma^{6}$
The Lennard-Jones Combining Rules
- The LJ interactions between different types of atoms are computed by combining the LJ parameters.
- Avoid a huge number of parameters for each combination of different atom types.
- Different force fields use different combining rules.

- The arithmetic mean (Lorentz) is motivated by collision of hard spheres
- The geometric mean (Berthelot) has little physical argument.
Geometric mean:
\(C12_{ij}=\sqrt{C12_{ii}\times{C12_{jj}}}\qquad C6_{ij}=\sqrt{C6_{ii}\times{C6_{jj}}}\qquad\) (GROMOS)
\(\sigma_{ij}=\sqrt{\sigma_{ii}\times\sigma_{jj}}\qquad\qquad\qquad \epsilon_{ij}=\sqrt{\epsilon_{ii}\times\epsilon_{jj}}\qquad\qquad\) (OPLS)
Lorentz–Berthelot:
\(\sigma_{ij}=\frac{\sigma_{ii}+\sigma_{jj}}{2},\qquad \epsilon_{ij}=\sqrt{\epsilon_{ii}\times\epsilon_{jj}}\qquad\) (CHARMM, AMBER).
- Known issues: overestimates the well depth
Less common combining rules.
Waldman–Hagler:
\(\sigma_{ij}=\left(\frac{\sigma_{ii}^{6}+\sigma_{jj}^{6}}{2}\right)^{\frac{1}{6}}\) , \(\epsilon_{ij}=\sqrt{\epsilon_{ii}\epsilon_{jj}}\times\frac{2\sigma_{ii}^3\sigma_{jj}^3}{\sigma_{ii}^6+\sigma_{jj}^6}\)
This combining rule was developed specifically for simulation of noble gases.
Hybrid (the Lorentz–Berthelot for H and the Waldman–Hagler for other elements). Implemented in the AMBER-ii force field for perfluoroalkanes, noble gases, and their mixtures with alkanes.
The Buckingham potential
- Replaces the repulsive \(r^{-12}\) term in Lennard-Jones potential with exponential function of distance:
$V_{B}(r)=Aexp(-Br) -\frac{C}{r^{6}}$

- Exponential function describes electron density more realistically
- Computationally more expensive to calculate.
- Risk of “buckingham catastrophe” at short distances.
There is only one combining rule for Buckingham potential in GROMACS:
$A_{ij}=\sqrt{A_{ii}\times{A_{jj}}}$
$B_{ij}=\frac{2}{\frac{1}{B_{ii}}+\frac{1}{B_{jj}}}$
$C_{ij}=\sqrt{C_{ii}\times{C_{jj}}}$
Combining rule (GROMACS):
\(A_{ij}=\sqrt{A_{ii}\times{A_{jj}}} \qquad B_{ij}=\frac{2}{\frac{1}{B_{ii}}+\frac{1}{B_{jj}}} \qquad C_{ij}=\sqrt{C_{ii}\times{C_{jj}}}\)
Specifying Combining Rules
GROMACS
Combining rule is specified in the [defaults] section of the forcefield.itp file (in the column ‘comb-rule’).
[ defaults ] ; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ 1 2 yes 0.5 0.8333Geometric mean is selected by using rules 1 and 3; Lorentz–Berthelot rule is selected using rule 2.
GROMOS force field requires rule 1; OPLS requires rule 3; CHARMM and AMBER require rule 2
The type of potential function is specified in the ‘nbfunc’ column: 1 selects Lennard-Jones potential, 2 selects Buckingham potential.
NAMD
By default, Lorentz–Berthelot rules are used. Geometric mean can be turned on in the run parameter file:
vdwGeometricSigma yes
The electrostatic potential
- Point charges are assigned to the positions of atomic nuclei to approximate the electrostatic potential around a molecule.
- The Coulomb’s law: $V_{Elec}=\frac{q_{i}q_{j}}{4\pi\epsilon_{0}\epsilon_{r}r_{ij}}$
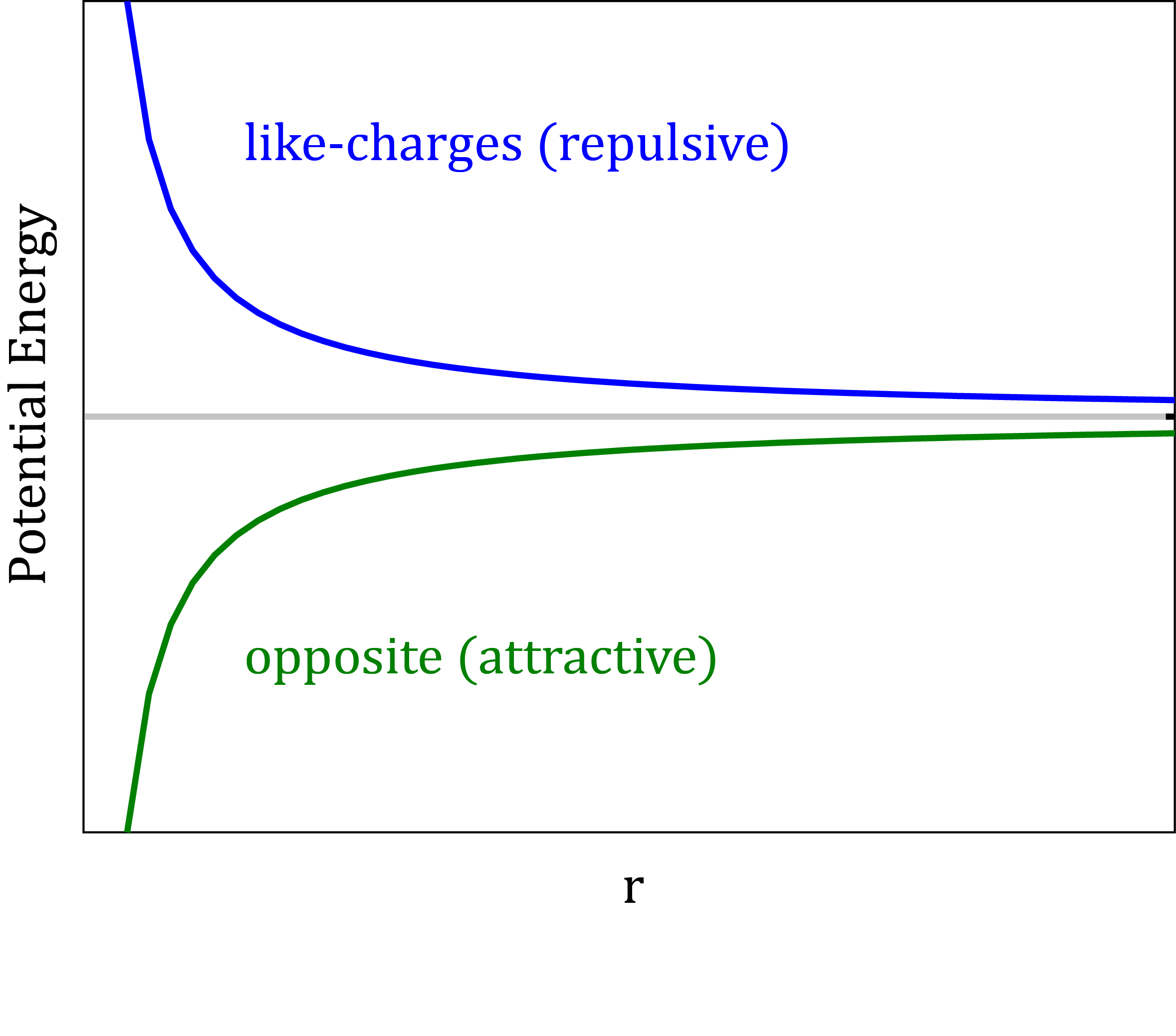
Electronic polarization
Types of polarizable force fields:
- Fluctuating charges: polarization is modeled as a charge transfer process between atoms. In recent years, has not been actively developed.
- Drude oscillators: massless charged particles attached to atoms via harmonic springs (CHARMM-Drude, OPLS5).
- Inducible point dipoles (AMOEBA).
- Gaussian Electrostatic Model (GEM). Gaussian Charge density + AMOEBA polarization.
- Polarizable Gaussian Multipole (pGM). Permanent Gaussian multipoles + Inducible Gaussian dipoles.
Short-range and Long-range Interactions
- Interaction is short-range if the potential decreases faster than r-3
- The Lennard-Jones interactions are short-ranged, r-6.
- The Coulomb interactions are long-ranged, r-1.
Bonded Terms
The bond potential
- Oscillation about an equilibrium bond length r0 with bond constant kb: $V_{Bond}=k_b(r_{ij}-r_0)^2$
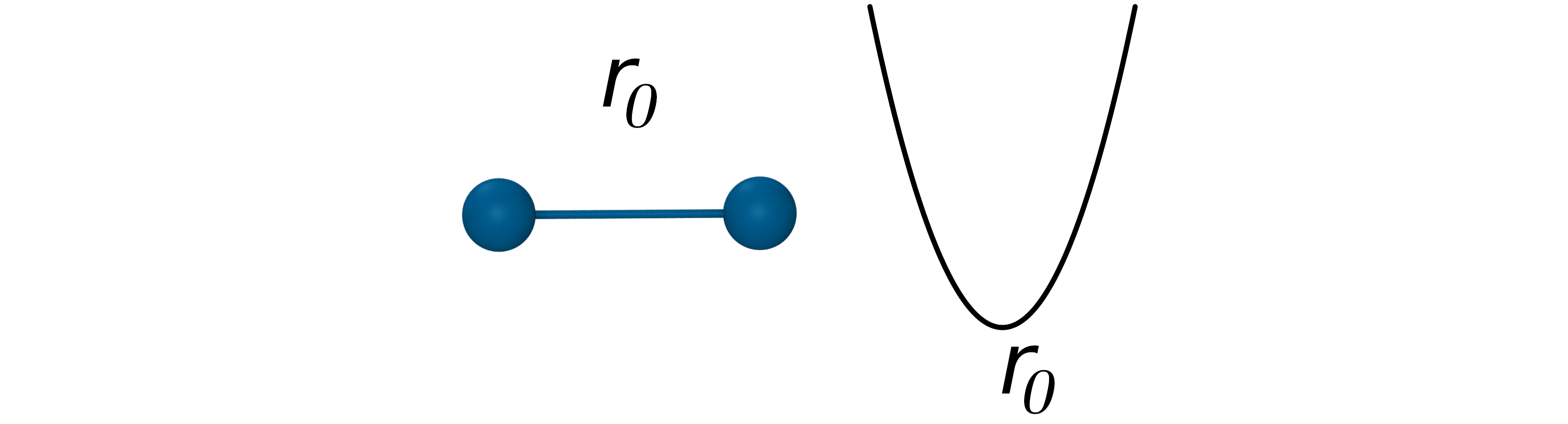
- Poor approximation at extreme stretching, but it works well at moderate temperatures.
The angle potential
- Oscillation about an equilibrium angle \(\theta_{0}\) with force constant \(k_\theta\): $V_{Angle}=k_\theta(\theta_{ijk}-\theta_0)^2$
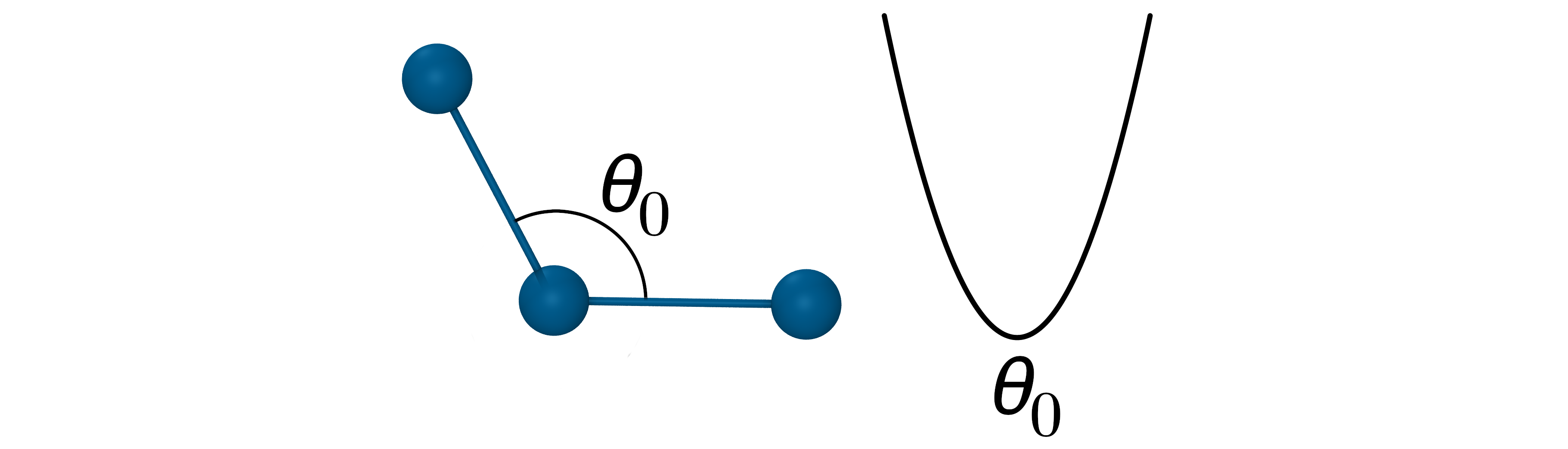
- The force constants for angle potential are about 5 times smaller that for bond stretching.
The torsion (dihedral) angle potential
- Defined for every 4 sequentially bonded atoms.
- Sum of any number of periodic functions, n - periodicity, \(\delta\) - phase shift angle.
$V_{Dihed}=k_\phi(1+cos(n\phi-\delta)) + …$
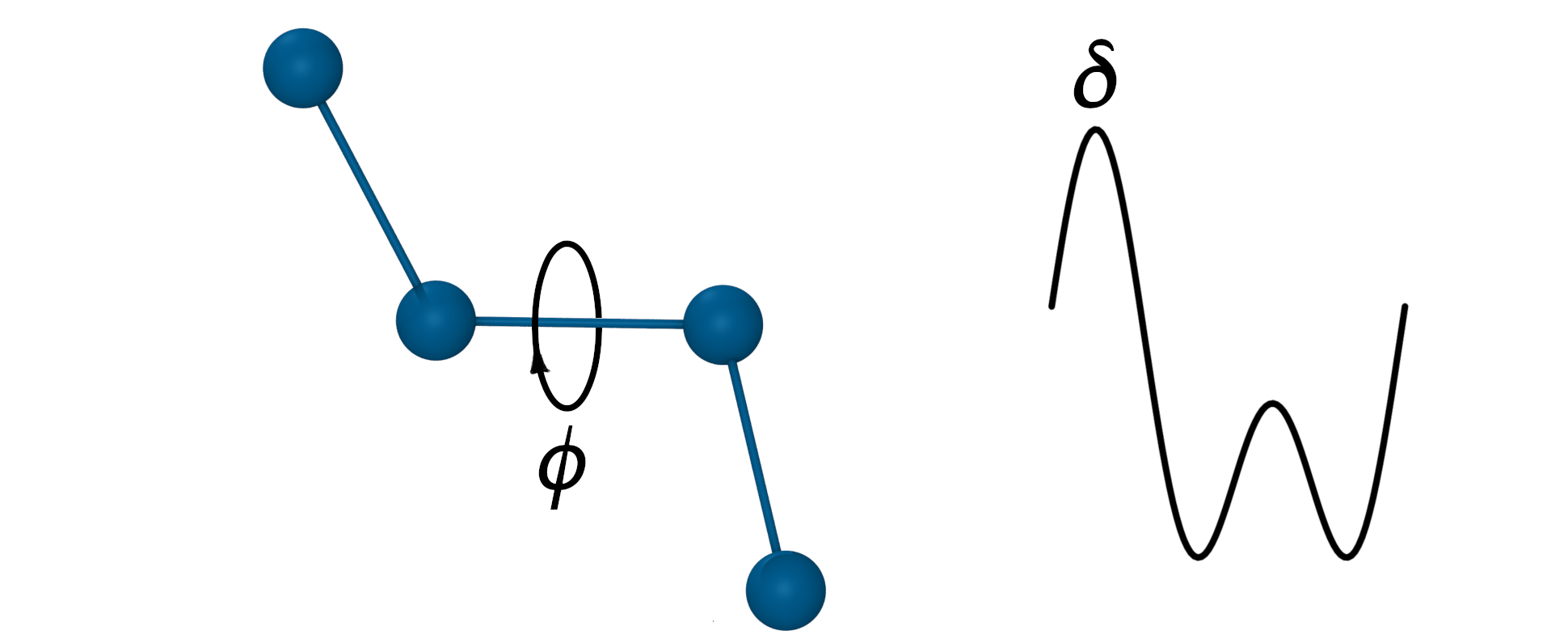
- n represents the number of potential maxima or minima generated in a 360° rotation.

- Combination of n=2 and n=3 dihedrals used to reproduce cis/trans and trans/gauche energy differences in ethylene glycol
The improper torsion potential
- Also known as ‘out-of-plane bending’
- Defined for a group of 4 bonded atoms where the central atom i is connected to the 3 peripheral atoms j,k, and l.
- Used to enforce planarity.
- Given by a harmonic function: $V_{Improper}=k_\phi(\phi-\phi_0)^2$

- The dihedral angle \(\phi\) is the angle between planes ijk and ijl.
Coupling Terms
The Urey-Bradley potential
- Coupling between bond length and bond angle is described by the Urey-Bradley potential.
- The Urey-Bradley term is defined as a spring between the outer atoms of a bonded triplet.
- Approximated by a harmonic function: $V_{UB}=k_{ub}(r_{jk}-r_{ub})^2$
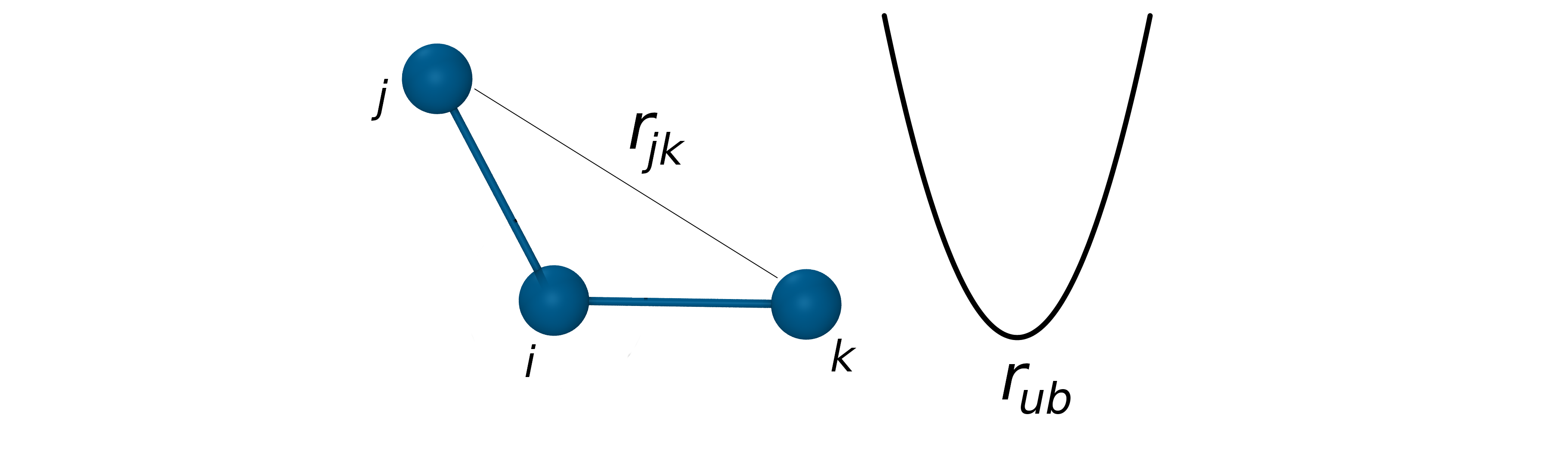
- Improve agreement with vibrational spectra.
- Do not affect overall conformational sampling.
- Implemented in CHARMM and AMOEBA force fields.
CHARMM CMAP correction potential
- Peptide torsion angles: $\phi$, $\psi$, $\omega$.
- A protein can be seen as a series of linked sequences of peptide units which can rotate around $\phi/\psi$ angles.
- $\phi/\psi$ angles define the conformation of the backbone.
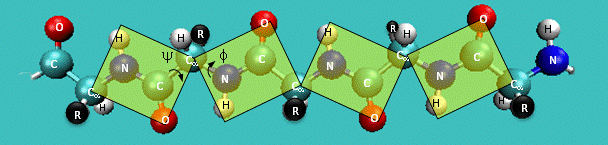
- $\phi/\psi$ dihedral angle potentials correct for force field deficiencies such as errors in non-bonded interactions, electrostatics, lack of coupling terms, inaccurate combination, etc.
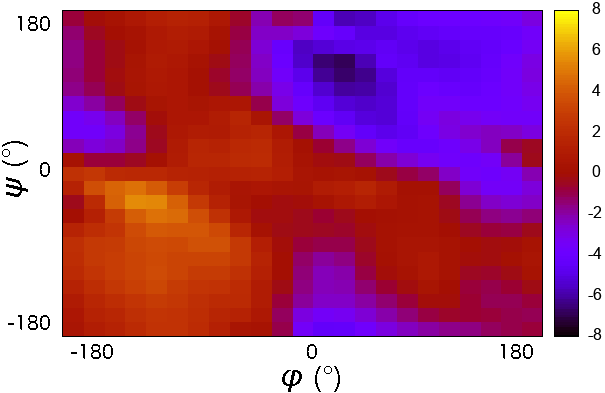
- CMAP potential was developed to improve the sampling of backbone conformations.
- CMAP parameter does not define a continuous function.
- It is a grid of energy correction factors defined for each pair of$\phi/\psi$ angles typically tabulated with 15 degree increments.
Energy scale of potential terms
\(k_BT\) at 298 K ~ 0.593 \(\frac{kcal}{mol}\) Bond vibrations ~ 100 ‐ 500 \(\frac{kcal}{mol \cdot \unicode{x212B}^2}\) Bond angle bending ~ 10 - 50 \(\frac{kcal}{mol \cdot deg^2}\) Dihedral rotations ~ 0 - 2.5 \(\frac{kcal}{mol \cdot deg^2}\) van der Waals ~ 0.5 \(\frac{kcal}{mol}\) Hydrogen bonds ~ 0.5 - 1.0 \(\frac{kcal}{mol}\) Salt bridges ~ 1.2 - 2.5 \(\frac{kcal}{mol}\)
Exclusions from Non-Bonded Interactions
- In pairs of atoms connected by chemical bonds bonded energy terms replace non-bonded interactions.
- All pairs of connected atoms separated by up to 2 bonds (1-2 and 1-3 pairs) are excluded from non-bonded interactions. It is assumed that they are properly described with bond and angle potentials.

- 1-4 interaction represents a special case where both bonded and non-bonded interactions are required for a reasonable description. However, due to the short distance between the 1–4 atoms full strength non-bonded interactions are too strong and must be scaled down.
- Non-bonded interaction between 1-4 pairs depends on the specific force field.
- Some force fields exclude VDW interactions and scale down electrostatic (AMBER) while others may modify both or use electrostatic as is.
The application of Machine Learning to MD simulations
- Classical force fields are inaccurate. They limit the predictive power of MD simulations.
- QM methods are accurate but require enormous computing power.
Could we calculate accurate potential energy from molecular structures without knowing their electronic structures?
What if we used a function that has no physical meaning but is computationally efficient and flexible enough to describe potential energy surfaces accurately?

- Potential energy is fully determined by a molecule’s geometry and atomic composition, so it is theoretically possible.
- Train a model on high level QM calculations
- Predict potential energy directly from an atomic structure.
What Information Can MD Simulations Provide?
With the help of MD it is possible to model phenomena that cannot be studied experimentally. For example
- Understand atomistic details of conformational changes, protein unfolding, interactions between proteins and drugs
- Study thermodynamics properties (free energies, binding energies)
- Study biological processes such as (enzyme catalysis, protein complex assembly, protein or RNA folding, etc).
For more examples of the types of information MD simulations can provide read the review article: Molecular Dynamics Simulation for All.
Challenge: Counting Non-Bonded Interactions
How many non-bonded interactions are in the system with ten Argon atoms? 10, 45, 90, or 200?
Solution
Argon atoms are neutral, so there is no Coulomb interaction. Atoms don’t interact with themselves and the interaction ij is the same as the interaction ji. Thus the total number of pairwise non-bonded interactions is (10x10 - 10)/2 = 45.
Challenge: Exclusions
How many 1-4 interactions between carbon atoms are in the system composed of 100 pentane (H3C-CH2-CH2-CH2-CH3) molecules?
Solution
200
Challenge: Interaction potentials
Which potential term describes the interaction between two molecules? CMAP, Urey-Bradley, Lennard-Jones or angle?
Solution
The Lennard-Jones potential
Specifying Exclusions
GROMACS
The exclusions are generated by grompp as specified in the [moleculetype] section of the molecular topology .top file:
[ moleculetype ] ; name nrexcl Urea 3 ... [ exclusions ] ; ai aj 1 2 1 3 1 4In the example above non-bonded interactions between atoms that are no farther than 3 bonds are excluded (nrexcl=3). Extra exclusions may be added explicitly in the [exclusions] section.
The scaling factors for 1-4 pairs, fudgeLJ and fudgeQQ, are specified in the [defaults] section of the forcefield.itp file. While fudgeLJ is used only when gen-pairs is set to ‘yes’, fudgeQQ is always used.
[ defaults ] ; nbfunc comb-rule gen-pairs fudgeLJ fudgeQQ 1 2 yes 0.5 0.8333NAMD
Which pairs of bonded atoms should be excluded is specified by the exclude parameter.
Acceptable values: none, 1-2, 1-3, 1-4, or scaled1-4exclude scaled1-4 1-4scaling 0.83 scnb 2.0If scaled1-4 is set, the electrostatic interactions for 1-4 pairs are multiplied by a constant factor specified by the 1-4scaling parameter. The LJ interactions for 1-4 pairs are divided by scnb.
Key Points
Molecular dynamics simulates atomic positions in time using forces acting between atoms
The forces in molecular dynamics are approximated by simple empirical functions