Fast Methods to Evaluate Forces
Overview
Teaching: 30 min
Exercises: 0 minQuestions
Why the computation of non-bonded interactions is the speed limiting factor?
How are non-bonded interactions computed efficiently?
What is a cutoff distance?
How to choose the appropriate cutoff distance?
Objectives
Understand how non-bonded interactions are truncated.
Understand how a subset of particles for calculation of short-range non-bonded is selected.
Learn how to choose the appropriate cutoff distance and truncation method.
Learn how to select cutoff distance and truncation method in GROMACS and NAMD.
Challenges in calculation of non bonded interactions.
Why Are Non-Bonded Interactions the Computational Bottleneck?
- The number of non-bonded interactions increases as the square of the number of atoms.
- The most computationally demanding part of a molecular dynamics simulation is the calculation of the non-bonded interactions.
- Simulation can be significantly accelerated by limiting the number of evaluated non bonded interactions.
- Exclude pairs of atoms separated by long distance.
- Maintain a list of all particles within a predefined cutoff distance of each other.
The Cutoff Distance
A cutoff distance defines the maximum separation at which a short-range interaction is explicitly calculated.
- using cutoff dramatically reduces the number of force calculations
How to identify which atoms are within the cutoff distance?
Neighbour Searching Methods
- Divide simulation system into grid cells - cell lists.
- Compile a list of neighbors for each particle by searching all pairs - Verlet lists.
Cell Lists
- Divide the simulation domain into cells with edge length greater or equal to the cutoff distance.
- Interaction potential of a particle is the sum of the pairwise interactions with all other particles in the same cell and all neighboring cells.
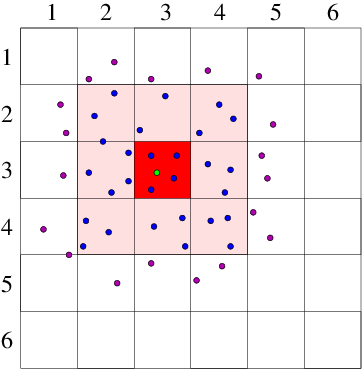
Verlet Lists
- Verlet list stores all particles within the cutoff distance plus some extra buffer distance.
- All pairwise distances must be evaluated.
- List is valid until any particle has moved more than half of the buffer distance.
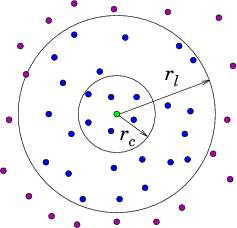
- Efficient computation of pairwise interactions.
- Relatively large memory requirement.
- In practice, almost all simulations use a combination of spatial decomposition and Verlet lists.
Truncation of the Electrostatic Interactions
Electrostatic interactions decay slowly with distance and can remain significant even between atoms that are far apart in the system.
- The electrostatic interaction is divided into two parts: a short-range and a long-range.
- The short-range contribution is calculated by exact summation.
- The forces beyond the cutoff radius are approximated using Particle-Mesh Ewald (PME) method.
Periodic boundary conditions provide the framework needed to handle long-range interactions.
Problems with Truncation of Lennard-Jones Interactions and How to Avoid Them?
- LJ potential is always truncated at the cutoff distance.
- Truncation introduces a discontinuity in the potential energy.
- A sharp change in potential may result in nearly infinite forces.
Methods to Handle Cutoff Artifacts
 Figure 1. The Distance Dependence of Potential and Force for Different Truncation Methods
Figure 1. The Distance Dependence of Potential and Force for Different Truncation Methods
| Shifted potential | |
$\circ$ Shift the whole potential uniformly by adding a constant at values below cutoff. $\circ$ Avoids infinite forces. $\circ$ Does not change forces at the distances below cutoff. $\circ$ Introduces a discontinuity in the force at the cutoff distance. $\circ$ Modifies total potential energy. |
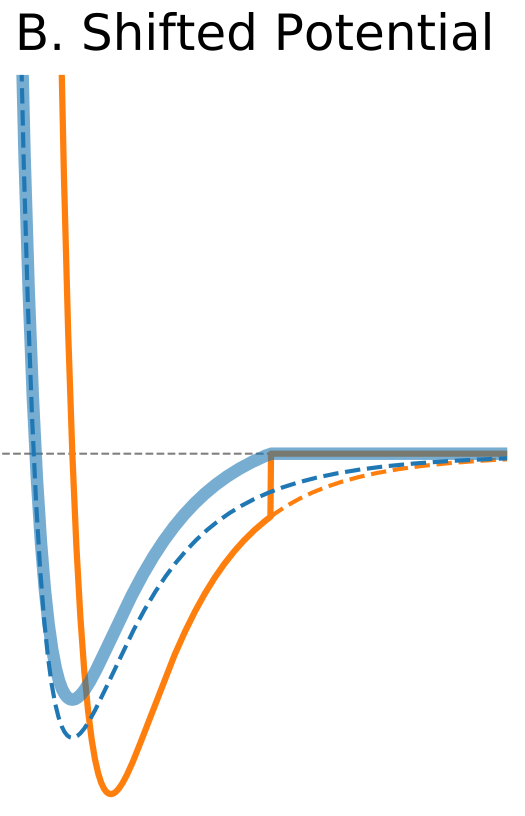 |
| Shifted Force | |
$\circ$ Shift the whole force so that it vanishes at the cutoff distance. $\circ$ Modifies equations of motion at all distances. $\circ$ Better results at shorter cutoff values compared to the potential shift. |
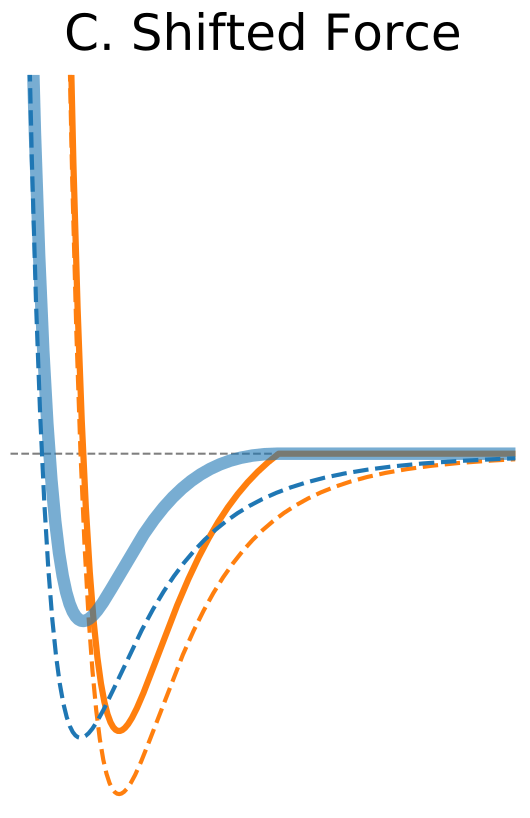 |
| Switching Function | |
$\circ$ Modify the shape of the potential function near cutoff. $\circ$ Forces are modified only near the cutoff boundary and they approach zero smoothly. |
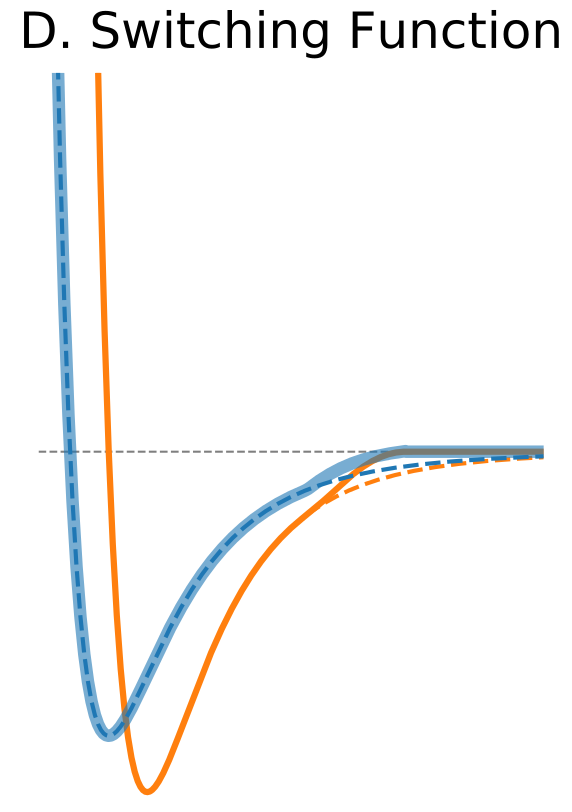 |
Which Method Should You Use?
Trade-offs between:
- physical accuracy
- numerical stability
- computational efficiency
How to Choose the Appropriate Cutoff Distance?
- A common practice is to truncate at 2.5 \(\sigma\).
- At this distance, the LJ potential is about 1/60 of the well depth \(\epsilon\).
- The choice of the cutoff distance depends on the force field and atom types.
For example for the O, N, C, S, and P atoms in the AMBER99 force field the values of \(\sigma\) are in the range 1.7-2.1, while for the Cs ions \(\sigma=3.4\). Thus the minimum acceptable cutoff, in this case, is 8.5.
Bigger Cutoffs Are Not Always Better
- Increasing cutoff does not necessarily improve accuracy.
- Each force field has been developed using a certain cutoff value, and effects of the truncation were compensated by adjustment of some other parameters.
- To ensure consistency and reproducibility of simulation you should choose the cutoff appropriate for the force field:
Table 1. Cutoffs Used in Development of the Common Force Fields
| AMBER | CHARMM | GROMOS | OPLS |
|---|---|---|---|
| 8 Å, 10 Å (ff19SB) | 12 Å | 14 Å | 11-15 Å (depending on a molecule size) |
Properties that are very sensitive to the choice of cutoff
- the surface tension (Ahmed, 2010),
- the solid–liquid coexistence line (Grosfils, 2009),
- the lipid bi-layer properties (Huang, 2014),
- the structural properties of proteins (Piana, 2012).
For such quantities even a cutoff at 2.5 \(\sigma\) gives inaccurate results, and in some cases the cutoff larger than 6 \(\sigma\) was required for reliable simulations (Grosfils, 2009).
Effect of cutoff on energy conservation
- Short cutoff may lead to an increase in the temperature of the system over time.
- The best practice is to carry out trial simulations without temperature control to test it for energy leaks or sources before a production run.
Challenge: Truncation of VDW Interactions
Which truncation method modifies VDW interactions only near the cut-off distance?
Solution
Switching functions
Selecting Neighbour Searching Methods
GROMACS
Neighbour searching is specified in the run parameter file mdp.
cutoff-scheme = group ; Generate a pair list for groups of atoms. Since version 5.1 group list has been deprecated and only Verlet scheme is available. cutoff-scheme = Verlet ; Generate a pair list with buffering. The buffer size is automatically set based on verlet-buffer-tolerance, unless this is set to -1, in which case rlist will is used. ; Neighbour search method. ns-type = grid ; Make a grid in the box and only check atoms in neighboring grid cells. ns-type = simple ; Loop over every atom in the box. nstlist = 5 ; Frequency to update the neighbour list. If set to 0 the neighbour list is constructed only once and never updated. The default value is 10.NAMD
When run in parallel NAMD uses a combination of spatial decomposition into grid cells (patches) and Verlet lists with extended cutoff distance.
stepspercycle 10 # Number of timesteps in each cycle. Each cycle represents the number of timesteps between atom reassignments. Default value is 20. pairlistsPerCycle 2 # How many times per cycle to regenerate pairlists. Default value is 2.
Selecting LJ Potential Truncation Method
GROMACS
Truncation of LJ potential is specified in the run parameter file mdp.
vdw-modifier = potential-shift ; Shifts the VDW potential by a constant such that it is zero at the rvdw. vdw-modifier = force-switch ; Smoothly switches the forces to zero between rvdw-switch and rvdw. vdw-modifier = potential-switch ; Smoothly switches the potential to zero between rvdw-switch and rvdw. vdw-modifier = none rvdw-switch = 1.0 ; Where to start switching. rvdw = 1.2 ; Cut-off distanceNAMD
Truncation of LJ potential is specified in the run parameter file mdin.
cutoff 12.0 # Cut-off distance common to both electrostatic and van der Waals calculations switching on # Turn switching on/off. The default value is off. switchdist 10.0 # Where to start switching vdwForceSwitching on # Use force switching for VDW. The default value is off.AMBER force fields
AMBER force fields are developed with hard truncation. Do not use switching or shifting with these force fields.
Selecting Cutoff Distance
GROMACS
Cutoff and neighbour searching is specified in the run parameter file mdp.
rlist = 1.0 ; Cutoff distance for the short-range neighbour list. Active when verlet-buffer-tolerance = -1, otherwise ignored. verlet-buffer-tolerance = 0.002 ; The maximum allowed error for pair interactions per particle caused by the Verlet buffer. To achieve the predefined tolerance the cutoff distance rlist is adjusted indirectly. To override this feature set the value to -1. The default value is 0.005 kJ/(mol ps).NAMD
When run in parallel NAMD uses a combination of spatial decomposition into grid cells (patches) and Verlet lists with extended cutoff distance.
pairlistdist 14.0 # Distance between pairs for inclusion in pair lists. Should be bigger or equal than the cutoff. The default value is cutoff. cutoff 12.0 # Local interaction distance. Same for both electrostatic and VDW interactions.
Key Points
The calculation of non-bonded forces is the most computationally demanding part of a molecular dynamics simulation.
Non-bonded interactions are truncated to speed up simulations.
The cutoff distance should be appropriate for the force field and the size of the periodic box.