Scripting
Overview
Teaching: 30 min
Exercises: 10 minQuestions
How to work with atom selections?
How to change attributes of selected atoms?
How to move and superimpose selections?
How to write loops
How to measure distances between groups of atoms?
Objectives
Learn to work with atom selections?
Learn to move and superimpose selections?
Learn to write loops in VMD
Learn to measure distances between groups of atoms?
Tcl Scripting in VMD
In addition to commands, VMD offers the built-in Tcl programming language. VMD Tcl scripts can help you investigate molecule properties and perform analysis.
Tcl language
Tcl is shortened form of Tool Command Language. This language combines scripting with an interpreter that gets embedded in the application. There are embedded Tcl interpreters in both VMD and NAMD. Tcl interpreter available from the command-line allows you to write data processing and visualization scripts utilizing any existing VMD functions and variables. For example, you have access to all information about loaded molecules such as coordinates, atom names, occupancy, and charge.
- Tcl interpreter is embedded in both VMD and NAMD.
Selecting atoms
Rotating or translating molecule that we have done so far simply changes viewpoint. It does not change coordinates of the loaded molecules. In your work you may want to rotate or translate a molecule and save its modified coordinates. It may be useful for example if you want to prepare a simulation system containing several copies of a molecule prepared from one structure file.
Many VMD commands operate on selected groups of atoms rather than just on whole molecules. After a selection has been created, it can be modified (rotated, translated), different properties, such as occupancy and beta factors, can be set, and the modified selection can be saved as a file.
- VMD commands operate on selected groups of atoms
As an example, let’s load 1si4.pdb and select protein backbone:
set sel [atomselect top "backbone"]
If command is successful VMD responds with a name of the created selection function:
atomselect0
In your case, the number of the function may be different, depending on how many selections have already been made.
With this command we created a function for selecting atoms, and set a variable sel to point to it. sel is just the name of this variable, you can use any name you wish. You can think of the variable sel as a shortcut to atomselect0. Thus, atomselect0 is equivalent to $sel.
Working with selections
What commands are available for atomselect0?
- To get help on commands available for the function
atomselect0typeatomselect0or$sel.
Try using some of the commands:
$sel num # Returns the number of atoms
$sel writepdb backbone.pdb # writeXXX where XXX is a known format
$sel writegro backbone.gro
Let’s make another selection using the same variable sel:
set sel [atomselect top "resname CYS and name CA"]
atomselect1
This command resets the variable sel. After this command is executed sel points to atomselect1. This can be verified by displaying the selection text:
$sel text
resname CYS and name CA
What happened to atomselect0?
As you may have thought, atomselect0 as well as any other previously created selections still exist:
atomselect list
Selections can be deleted using the delete command.
atomselect0 delete
Changing properties of selected atoms
We have selected CA atoms of all CYS residues of the protein. Let’s do something useful with this selection:
Find out how may CYS residues are in our protein and what are their ID numbers:
set cys [$sel get resid] # make a list of CYS resid numbers
llength $cys # count the number of elements in the list
We can rename selected atoms:
$sel set name C1A
$sel get name
We can manipulate any other attribute of the selection!
Viewing selections
To show a selection in graphical window we first need to choose a drawing method for a new representation and then add the new representation to the molecule:
mol selection "resname CYS"
mol representation vdw
mol addrep top
Moving selected atoms
Translation
$sel moveby {0 50 0}
Complex transformations (rotate + translate)
The move command takes as an argument a [4x4] transformation matrix and applies it to the coordinates of each atom in the selection. A transformation matrix is computed from [3x3] rotation matrix and a translation vector. It is not straightforward to obtain it by hand. VMD has various commands to help creating transformation matrices.
- The
movecommand takes as an argument a [4x4] transformation matrix
The main command for generating transformation matrices is trans. It can create matrices for many transformations such as:
- centering a molecule
- adding an offset
- rotation about any axis {x y z}
- rotation about about a vector
- rotation about a bond
Click here for more details about matrix routines.
Let’s consider examples of some useful transformations.
Rotation around an axis
Rotate a molecule around x-axis by 30 degrees:
set sel [atomselect top all]
$sel move [trans axis x 30]
End of workshop 2024 - 3 hours
Rotation around a bond
- Convert cis-lutein to trans- by rotating around the bond C15-C35 by 180 degrees.
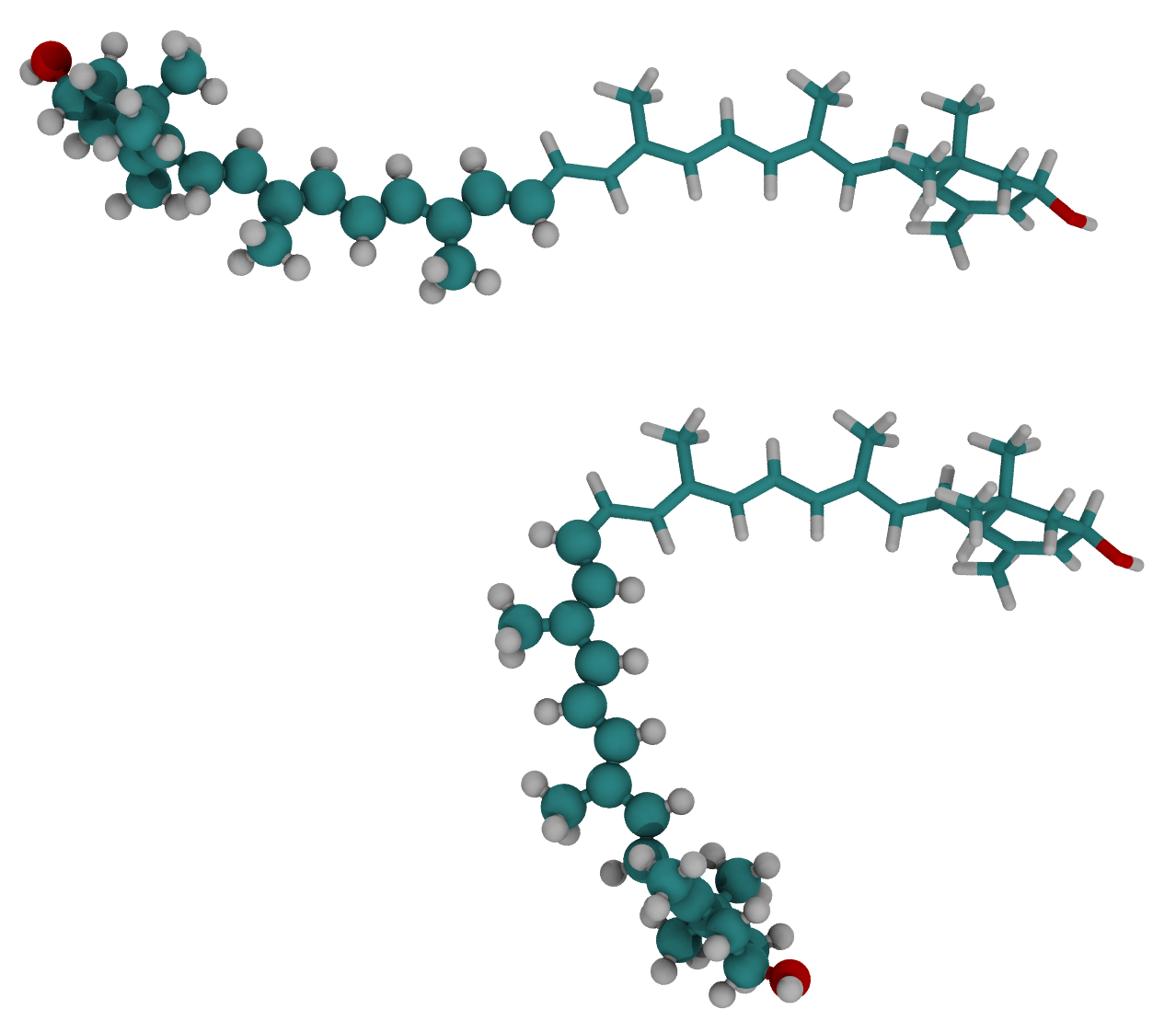
Load the file workshop_vmd/example_05/LUT-noh.pdb
# Select atoms to rotate
set sel [atomselect top "serial 1 to 21"]
# Select rotation axis atoms
set A1 [atomselect top "name C15"]
set A2 [atomselect top "name C35"]
# Get coordinates of these atoms
set AC1 [lindex [$A1 get {x y z}] 0]
set AC2 [lindex [$A2 get {x y z}] 0]
# Rotate selection
$sel move [trans bond $AC1 $AC2 180 deg]
lindex is a builtin Tcl command. It retrieves an element from a list.
Superposing molecules (more challenging)
Although VMD has GUI tool for aligning molecules, it is limited for molecules where lists of equivalent atoms can be constructed using the same selections command. This means that molecules must have either same atom names or same order of atoms. Often this is not the case. For example you may want to align several structurally similar ligands, or two homologous proteins that do not have exactly the same sequence.
With Tcl commands you can construct two independent lists of equivalent atoms and then use them for aligning molecules. The command for generating transformation matrices is measure fit. It creates transformation matrix for aligning two atom selections.
- How to align structurally similar molecules that do not have exactly the same atoms?
As an example, let’s consider two carotenoid molecules: lutein LUT and lycopene LYC.
PDB files of these molecules are in the directory workshop/pdb/carotenoids/.
The following code assumes that ID of the reference molecule is 0 and ID of the molecule that we want to move is 1, so make sure you use the right molecule IDs.
As these molecules have same polyene chains, but different end groups we will use middle part of molecules to align them.
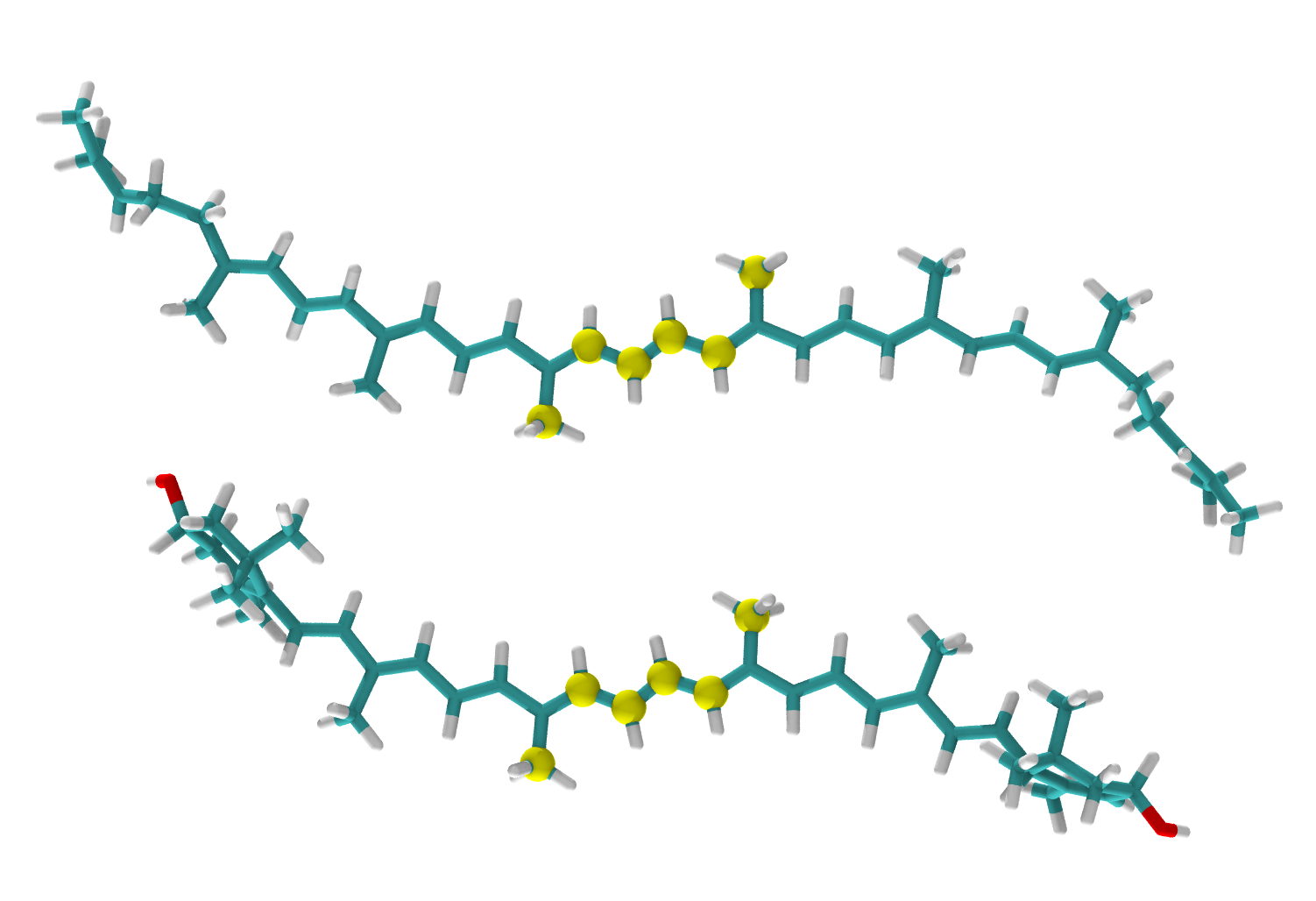
Atoms in these two pdb files are not in the same order, so we need to reorder them.
Selections do not depend on the order in which you list them in the selection command. Selections follow the order in which atoms appear in input files. To solve this problem you need to provide the list describing the order in which the reference atoms should be used to match the fit molecule.
Follow the following steps:
- list atoms in the fit molecule in the order of their index.
- make the list of matching atoms of the reference molecule
- make the list describing the order in which the reference atoms should be used to match the fit molecule
Example:
fitmol C18 C19 C20 C21 C50 C52
refmol C20 C14 C15 C35 C34 C40
order 2 0 1 4 3 5
set fitmol [atomselect 1 "name C18 C19 C20 C21 C50 C52"]
set refmol [atomselect 0 "name C20 C14 C15 C35 C34 C40"]
set trans_mat [measure fit $fitmol $refmol order {2 0 1 4 3 5}]
set allAtoms [atomselect 1 all]
$allAtoms move $trans_mat
Click here for more details
Loops in VMD scripts
Let’s write a simple for loop that animates zooming out.
A loop includes three statement:
- initialization of a loop variable
- termination condition
- increment
There statements are followed by a block of code that is executed repeatedly for each value of the loop variable.
for {set i 0} {$i < 200} {incr i} {scale by 0.99; display update}
Animate cis-trans transition of a lutein molecule
Create a loop that animates lutein’s cis-trans transition. You can use the rotation code that we developed above.
Making input files for NAMD.
NAMD uses beta and occupancy fields of PDB files as an input for various types of calculations. For example, such files are used to define
- position restraint parameters (restraint reference positions and force constant values for each atom)
- temperature coupling parameters (temperature coupling coefficient for each atom).
- constant forces
- collective variables
mol pdbload 1si4
set selAll [atomselect 0 all]
$selAll set occupancy 0
set selBackbone [atomselect 0 "protein and backbone"]
$selBackbone set occupancy 2.0
set selHEM [atomselect 0 "resname HEM"]
$selHEM set occupancy 10.0
$selAll writepdb "constraints.pdb"
Measuring distances between atoms vs. time
Measuring distance between a pair of atoms
Go to the directory with example MD data:
cd ~/scratch/workshop_vmd/example_02
This is a simulation of argonaute protein complexed with microRNA. As an example, let’s measure the distance between some RNA phosphate atoms and sodium ions attached to them.
The following are some pairs you might want to consider:
C895:P, Na+904:Na+
C890:P, Na+1136:Na+
C886:P, Na+1136:Na+
A883:P, Na+966:Na+
mol new prmtop_nowat.parm7
mol addfile mdcrd_nowat.xtc step 5 waitfor all
set file [open "distance.csv" w]
puts $file "Time (ns), Distance (A)"
set nf [molinfo top get numframes]
set sel1 [atomselect top "resid 895 and name P"]
set sel2 [atomselect top "resid 904"]
set bondList [measure bond "[$sel1 get index] [$sel2 get index]" first 0 last $nf]
for {set i 0} {$i < $nf} {incr i} {
set dist [lindex $bondList $i]
set time [expr $i/1000.0]
puts $file "$time, $dist"
}
close $file
Plotting data with Gnuplot
module load gnuplot
Start Gnuplot by typing gnuplot. Then in gnuplot command prompt enter the following commands:
set xlabel "Time (ns)"
set ylabel "Distance, (A)"
plot "distance.csv" skip 1 with lines
Measuring distances between groups of atoms
Measure distance between centers of mass of protein and nucleic acids
measure center <selection>- compute coordinates of the center of massvecsub <vec2> <vec1>- find vector connecting two centers of massveclength <vector>- compute distance
mol new prmtop_nowat.parm7
mol addfile mdcrd_nowat.xtc step 5 waitfor all
set file [open "distance.csv" w]
puts $file "Time (ns), Distance (A)"
set nf [molinfo top get numframes]
set prot [atomselect top "noh protein"]
set nucl [atomselect top "noh nucleic"]
for {set i 0} {$i < $nf} {incr i} {
$prot frame $i
$nucl frame $i
set prot_center [measure center $prot weight mass]
set nucl_center [measure center $nucl weight mass]
set dist [veclength [vecsub $prot_center $nucl_center]]
set time [expr $i/1000.0]
puts $file "$time, $dist"
}
close $file
Getting help on text commands
You can get help with text commands in several ways:
- This link provides a summary of basic text commands in VMD.
- You can find a detailed description of all commands, as well as their syntax in the online VMD manual.
- Help on commands for atom selections. Type an atom selection function to see a list of commands for it, e.g.
atomselect0. - Builtin Tcl commands: https://www.tcl.tk/man/tcl8.6/TclCmd/contents.html
Useful links: Using the atomselect command
Key Points